
Les LGM se caractérisent par une augmentation de la perméabilité de la barrière de filtration glomérulaire, entraînant une perte protéique prédominée par l’albumine. L’interaction synergique entre la dysrégulation immunitaire et les altérations podocytaires compromet l’intégrité de la membrane basale glomérulaire, conduisant à une protéinurie massive.
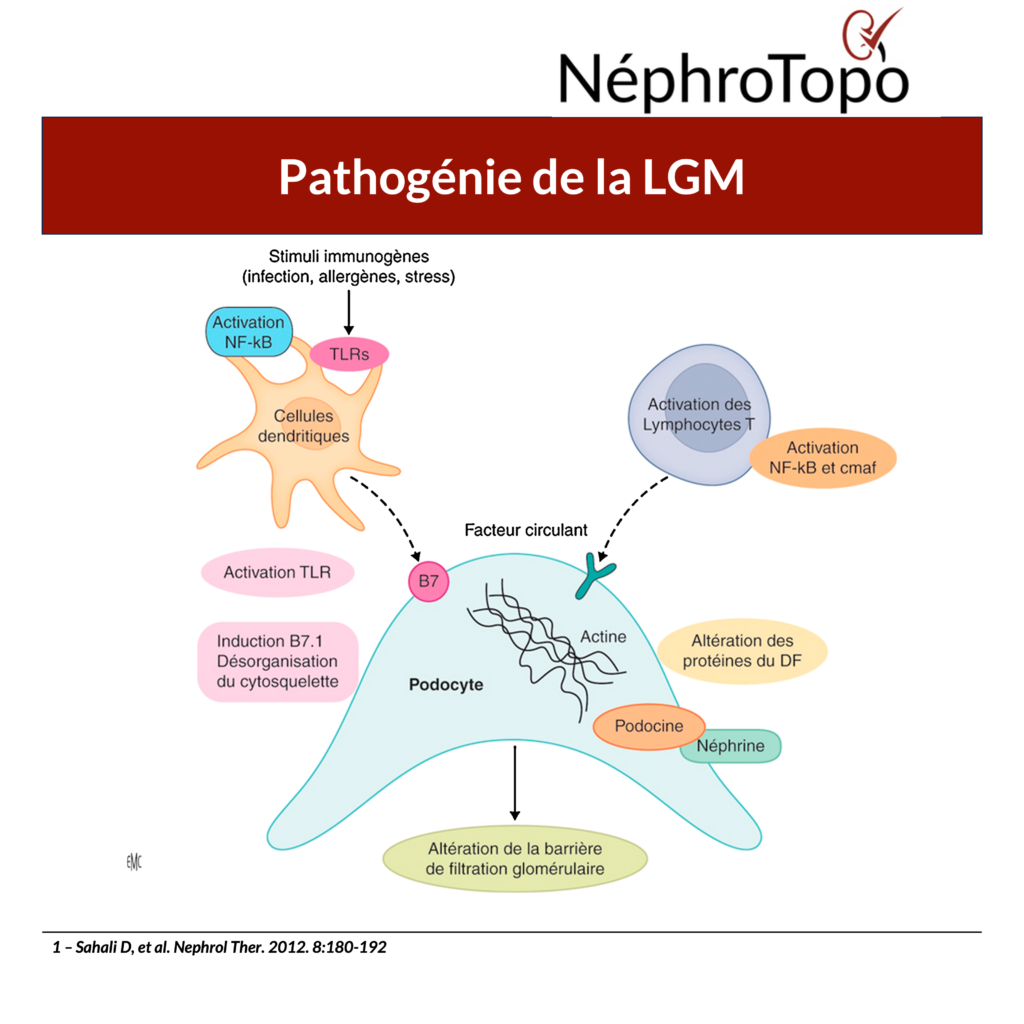
Dysrégulation immunitaire conduisant à la lésion podocytaire
La pathogénie des LGM implique un dérèglement du système immunitaire, en particulier un dysfonctionnement des lymphocytes T et B, qui conduit à la libération de facteurs circulants provoquant une lésion directe des podocytes, un effacement des pédicelles et une rupture de la barrière de filtration glomérulaire.
Les mécanismes immunitaires sont :
- Le dysfonctionnement des lymphocytes T est un élément central de la pathogenèse des LGM, l’activation anormale des lymphocytes T étant supposée libérer des facteurs de perméabilité circulants qui ciblent directement les podocytes.
- L’implication des lymphocytes B est de plus en plus reconnue, notamment compte tenu de la réponse thérapeutique au rituximab (anticorps anti-CD20), ce qui suggère que les lymphocytes B peuvent activer les lymphocytes T ou libérer des auto-anticorps dirigés contre la néphrine.
Les LGM sont caractérisées par l’absence de modifications inflammatoires ou de dépôts de complexes immuns dans le tissu rénal, ce qui la distingue des autres glomérulopathies et soutient l’hypothèse du facteur circulant.

Cytokines et pathogenèse des LGM
- Altération de la perméabilité glomérulaire : Certaines cytokines, en particulier IL-13, modifient l’expression des gènes des podocytes, ce qui entraîne (i) une perte ou un dysfonctionnement de la néphrine, (ii) une fragilisation de la barrière de filtration glomérulaire, entraînant une fuite protéique dans l’urine (iii) une activation des podocytes stressés, favorisant l’apoptose ou la dédifférenciation des podocytes.
Activation des cellules immunitaires et production d’auto-anticorps : L’IL-13 et d’autres cytokines comme IL-4 favorisent la différenciation des lymphocytes T auxiliaires de type 2 (Th2). Ces Th2 stimulent les lymphocytes B, qui peuvent produire des auto-anticorps dirigés contre la néphrine, exacerbant la lésion podocytaire. La présence de ces auto-anticorps contribue à l’auto-immunité locale et à la perte de protéines urinaires.
Promotion d’un environnement pro-inflammatoire : Les cytokines telles que TNF-α et IL-33 créent un milieu inflammatoire local, renforçant le stress podocytaire. L’IL-33/ST2 active les cellules immunitaires innées et Th2, qui sécrètent à leur tour IL-13, amplifiant la cascade inflammatoire et la protéinurie.

Cascade de lésions des podocytes
L’effacement des pédicelles des podocytes est la caractéristique pathologique visible en microscopie électronique, tandis qu’en microscopie optique, l’aspect est normal.
On observe au cours des LGM une diminution de l’expression des protéines podocytaires essentielles, notamment :
- Protéines du diaphragme de fente : néphrine, podocine, CD2AP et NEPH1 ;
- Protéine cytosquelettique : synaptopodine (l’expression est corrélée à la sensibilité aux stéroïdes).
L’intégrité structurale de la néphrine est vitale pour le maintien de la perméabilité sélective de la barrière de filtration glomérulaire. La redistribution ou la diminution de l’expression de la néphrine est fortement associée à l’apparition d’une protéinurie. De même, la podocine, le CD2AP et le NEPH1, qui contribuent à la stabilité du diaphragme de fente, peuvent aggraver la progression de la maladie lorsqu’elles sont dysfonctionnelles.
La surexpression de protéines pathogènes des podocytes contribue aux lésions :
- CD80 (B7-1) est une molécule co-stimulatrice des cellules présentatrices d’antigènes exprimée anormalement sur les podocytes dans les LGM ; l’activation des voies NF-κB par des antigènes externes induit une surexpression de CD80, provoquant des lésions du cytosquelette et une protéinurie.
- La surexpression de l’Angiopoïétine-like-4 (Angpti-4) : endommage la barrière de charge de la membrane basale glomérulaire et induit la fusion des pédicelles mais non spécifiques aux LGM.
Le CD80 est généralement exprimé à la surface des cellules immunitaires activées, mais son expression anormale dans les podocytes a été associée à la pathogenèse de la maladie. Des taux élevés de CD80 dans l’urine ou à la surface des podocytes lors des phases actives de la maladie suggèrent qu’un dérèglement immunitaire joue un rôle crucial dans les LGM.
Susceptibilité génétique
La prédisposition génétique joue un rôle crucial dans les LGM, notamment en déterminant le risque individuel de développer la maladie et la réponse au traitement. Les mutations de gènes clés tels que NPHS1 et NPHS2, qui codent respectivement pour les protéines néphrine et podocine, sont des facteurs bien documentés de la pathogenèse. D’autres gènes sont également impliqués, tels que WT1 et ACTN4.

La présence de ces mutations génétiques est souvent corrélée à des phénotypes plus sévères, à un début précoce et à une plus grande probabilité de résistance aux traitements conventionnels. De plus, les polymorphismes génétiques des gènes impliqués dans la production de cytokines, comme l’IL-13 et le TNF-alpha, peuvent moduler l’intensité de l’activation immunitaire, complexifiant davantage le traitement.
Corrélation clinico-pathologique
- La sensibilité aux cortocistéroïdes de la plupart des cas de LGM (70 à 90 % chez les enfants de plus d’un an) soutient l’hypothèse d’une pathogenèse à médiation immunitaire.
- L’évolution récurrente par rechutes et rémissions est compatible avec une activation immunitaire épisodique plutôt qu’avec des défauts structurels fixes.
- Les formes résistantes aux stéroïdes peuvent représenter une HSF précoce ou un mécanisme pathogène distinct, car une néphropathie à LGM non résolue peut évoluer vers une glomérulosclérose segmentaire et focale.
Journal |
